Flexible, once-daily oral dosing with BALVERSA®1
Multiple dosage strengths allow for flexible dosing
- Starting dose of BALVERSA® is 8 mg (two 4 mg tablets)
- BALVERSA® is taken orally once daily with or without food
- A dose increase to 9 mg (three 3 mg tablets) once daily is recommended based on tolerability, including hyperphosphatemia, at day 14 to 21 days
− Increase dose if serum phosphate level is <9.0 mg/dL and there are no ocular disorders or grade ≥2 adverse reactions
− If the phosphate level is ≥9.0 mg/dL, follow the relevant dose modifications listed in
Table 1: Dose modifications for hyperphosphatemia and soft tissue mineralization
The starting dose of BALVERSA® is 8 mg orally once daily1

No fasting restrictions. Tablets can be taken with or without food
![]()
Tablets should be swallowed whole

Missed doses should be taken as soon as possible on the same day and the regular daily dose taken on the next day; extra tablets should not be taken to make up for the missed dose
Flexible dosing allows for dose modifications as needed1
For full Dosing and Dose Modification, please refer to section 2 of the full BALVERSA® Prescribing Information.

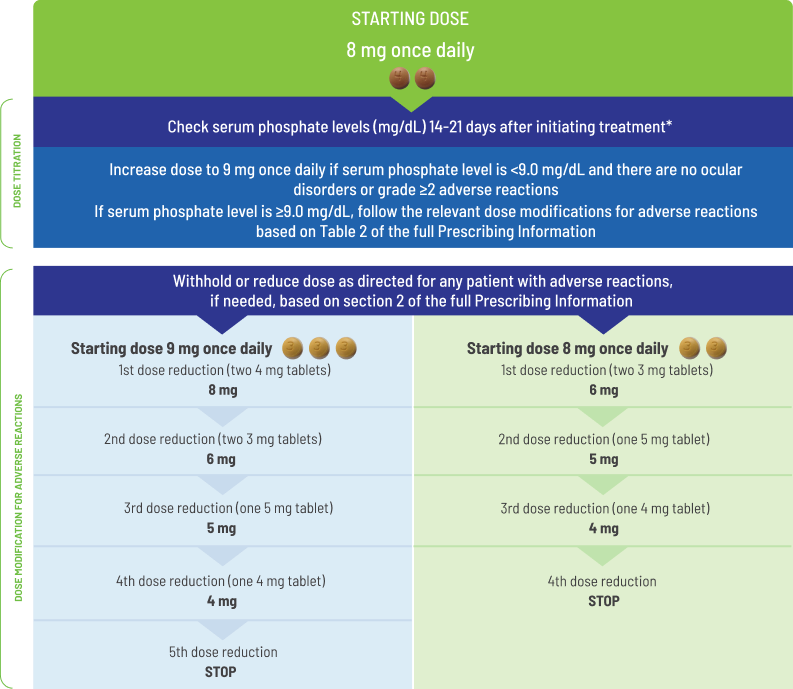
The recommended starting dose of BALVERSA® is 8 mg (two 4 mg tablets) orally once daily, with a dose increase to 9 mg (three 3 mg tablets) once daily based on tolerability, including hyperphosphatemia, at day 14 to 21 days. Treatment should continue until disease progression or unacceptable toxicity occurs.
Dose modifications and drug interactions1
For coadministration with moderate CYP2C9 or strong CYP3A4 inhibitors, consider alternative agents or monitor closely for adverse reactions.
Avoid concomitant use of strong CYP3A4 inducers with BALVERSA®.
For coadministration with moderate CYP3A4 inducers, increase the BALVERSA® dose up to 9 mg.
Serum phosphate level-altering agents: Avoid concomitant use with agents that can alter serum phosphate levels before the initial dose modification period.
P-gp substrates: Separate BALVERSA® administration by at least 6 hours before or after administration of P-gp substrates with narrow therapeutic index.
Managing hyperphosphatemia and soft tissue mineralization1
BALVERSA® can cause hyperphosphatemia leading to soft tissue mineralization, cutaneous calcinosis, non-uremic calciphylaxis and vascular calcification. Increases in phosphate levels are a pharmacodynamic effect of BALVERSA®. In the pooled safety population, increased phosphate occurred in 73% of BALVERSA®-treated patients. The median onset time of increased phosphate was 16 days (range: 8-421) after initiating BALVERSA®. Twenty-four percent of patients received phosphate binders during treatment with BALVERSA®. Vascular calcification was observed in 0.2% of patients treated with BALVERSA®.
Monitor for hyperphosphatemia throughout treatment. If serum phosphate is above 7 mg/dL, consider adding an oral phosphate binder until serum phosphate level returns to <7 mg/dL. Withhold, dose reduce, or permanently discontinue BALVERSA® based on duration and severity of hyperphosphatemia according to Table 1: Dose modifications for hyperphosphatemia and soft tissue mineralization.
Assess serum phosphate levels 14 to 21 days after initiating treatment. Increase the dose of BALVERSA® to 9 mg once daily if serum phosphate level is <9.0 mg/dL and there are no ocular disorders or Grade ≥2 adverse reactions. If the phosphate level is ≥9.0 mg/dL, follow the relevant dose modifications in Table 1: Dose modifications for hyperphosphatemia and soft tissue mineralization below. Monitor phosphate levels monthly for hyperphosphatemia.
Table 1: Dose modifications for hyperphosphatemia and soft tissue mineralization1
| Adverse Reaction | BALVERSA® Dose Modification |
|---|---|
| Hyperphosphatemia | |
In all patients, restrict phosphate intake to 600-800 mg daily. | |
<6.99 mg/dL | Continue BALVERSA® at current dose. |
7-8.99 mg/dL |
|
9-10 mg/dL |
|
>10 mg/dL |
|
Serum phosphate with life-threatening consequences; urgent intervention indicated (e.g., dialysis) | Discontinue BALVERSA® permanently. |
Please see full BALVERSA® Prescribing Information for dose modifications for elevated phosphate levels.
Managing eye disorders1
Perform monthly ophthalmological examinations during the first 4 months of treatment and every 3 months afterwards, and urgently at any time for visual symptoms. Ophthalmological examination should include assessment of visual acuity, slit lamp examination, fundoscopy, and optical coherence tomography. Visit the American Optometric Association website to refer your patient to an eye specialist.
Withhold BALVERSA® when central serous retinopathy (CSR) occurs and permanently discontinue if it does not resolve within 4 weeks or if Grade 4 severity.
Dose modifications for CSR1
For CRS of any grade, withhold BALVERSA® and perform an ophthalmic evaluation within 2 weeks:
- If improving within 14 days, restart BALVERSA® at the current dose
- If not improving within 14 days, withhold BALVERSA® until improving; once improving, may resume at the next lower dose level.
Upon restarting BALVERSA®, monitor for recurrence every 1 to 2 weeks for a month. If recurs or has not improved after 4 weeks of withholding BALVERSA®, consider permanent discontinuation.
Please see Safety and full BALVERSA® Prescribing Information for additional recommendations on managing potential eye disorders.
Managing other adverse reactions1
Table 2: Dose modifications for other adverse reactions1†
| Adverse Reaction | BALVERSA® Dose Modification |
|---|---|
Grade 3 | Withhold BALVERSA® until resolves to Grade 1 or baseline, then may resume dose level lower. |
Grade 4 | Permanently discontinue. |
†Dose adjustment graded using the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAEv4.03).
For detailed information on the management of adverse reactions, please see the full BALVERSA® Prescribing Information.
Reference
1. BALVERSA® [Prescribing Information]. Horsham, PA: Janssen Biotech, Inc.