The overall survival (OS) of BALVERSA® was evaluated in Cohort 1 of the phase 3 confirmatory THOR trial1
BALVERSA® was studied against chemotherapy* in a phase 3, randomized, open-label, multicenter clinical trial to evaluate OS of patients with advanced UC with susceptible selected FGFR alterations/mutations, who progressed after 1-2 prior treatments1
BALVERSA® study design1
The efficacy of BALVERSA® was evaluated in the THOR trial, a randomized, open-label, multicenter study in 266 patients with advanced urothelial cancer harboring selected FGFR3 alterations. All patients needed to have had disease progression after 1-2 prior treatments, at least 1 of which included a PD-1 or PD-L1 inhibitor. FGFR3 genetic alterations were identified from tumor tissue in a central laboratory by the QIAGEN therascreen ® FGFR RGQ RT-PCR kit in 75% of patients and 25% were identified by local NGS assays.
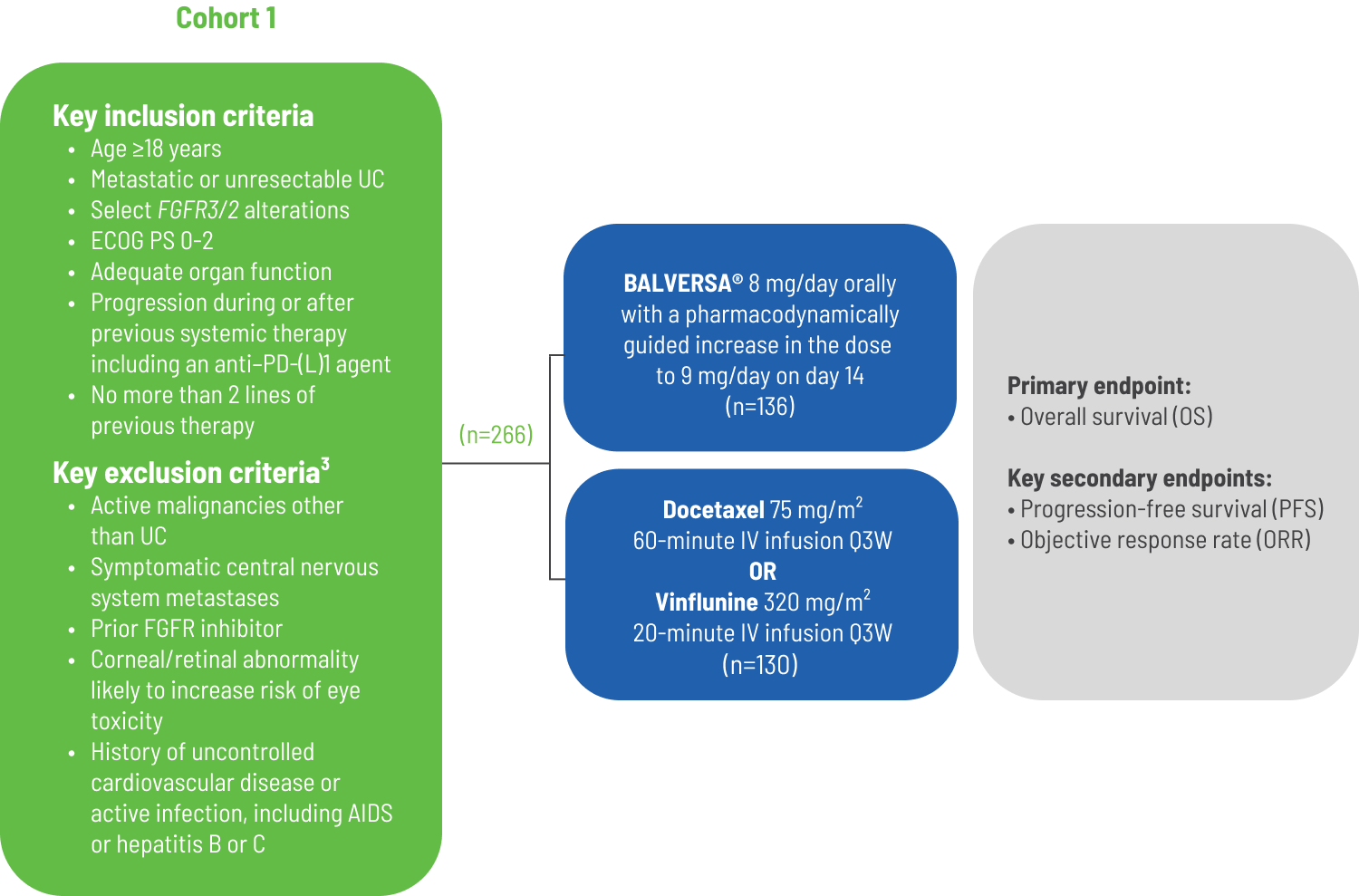
- Major efficacy outcome measures as determined by investigators according to RECIST v1.1 were1:
- primary endpoint:
- overall survival
- secondary endpoints:
- progression-free survival (PFS)
- objective response rate (ORR)
- duration of response (DoR)
- primary endpoint:
- Treatment was administered until unacceptable toxicity or progression
100% of patients in Cohort 1 with FGFR alterations had at least 1 FGFR3 alteration1,2
No patients had FGFR2 alterations.
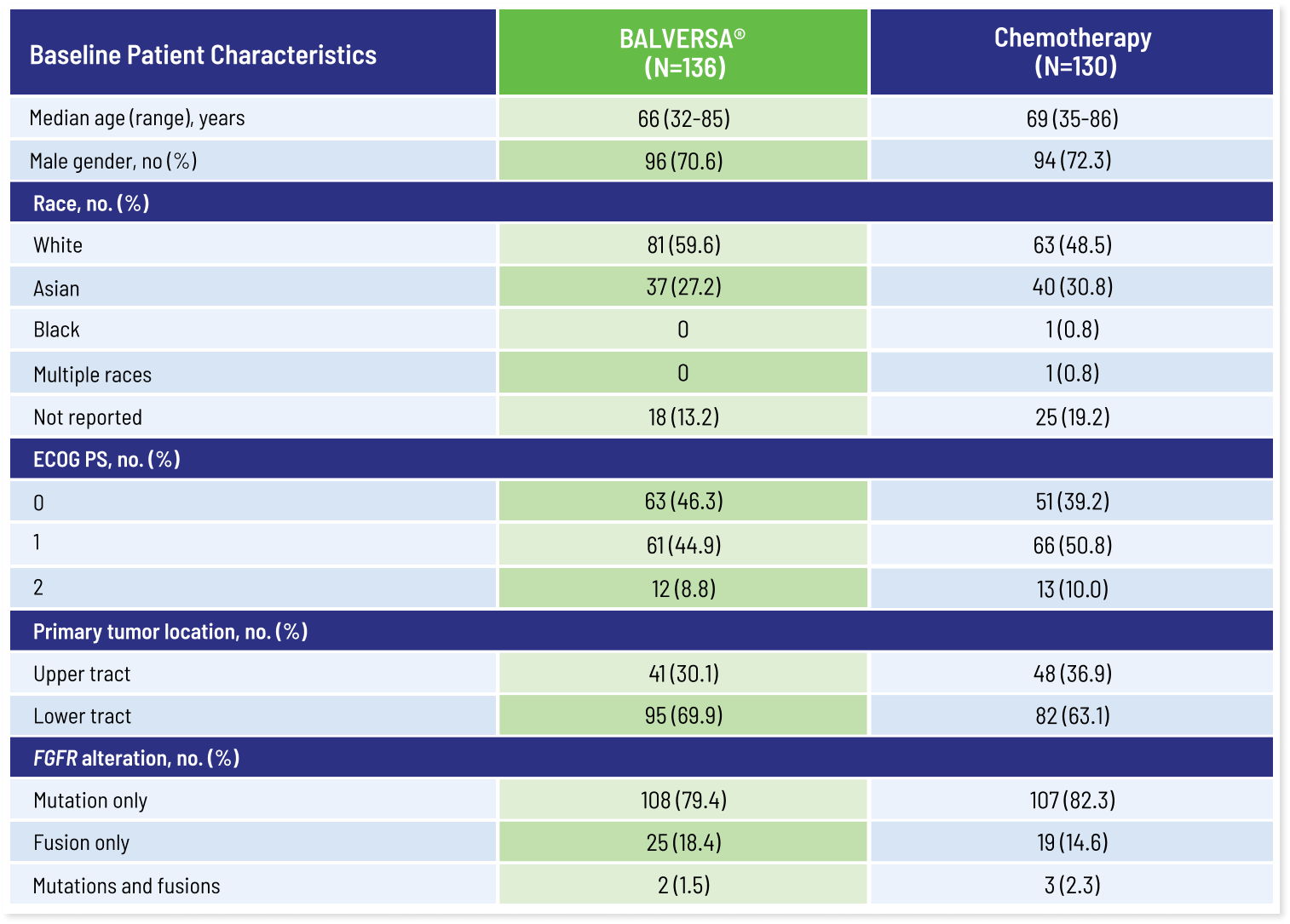
All patients needed to have had disease progression after 1 or 2 prior treatments, at least 1 of which included a PD-1 or PD-L1 inhibitor. Eighty-eight percent of patients received platinum-containing chemotherapy previously.
*Chemotherapy treatment options included docetaxel 75 mg/m2 once every 3 weeks or vinflunine 320 mg/m2 once every 3 weeks.
ECOG PS = Eastern Cooperative Oncology Group Performance Status; FGFR = fibroblast growth factor receptor; NGS = next-generation sequencing; PCR = polymerase chain reaction; PD-L1/PD-1 = programmed death ligand 1/programmed cell death protein 1; Q3W = every three weeks; RECIST = Response Evaluation Criteria In Solid Tumors; UC = urothelial carcinoma.
References
1. BALVERSA® [Prescribing Information]. Horsham, PA: Janssen Biotech, Inc. 2. Loriot Y, Matsubara N, Park SH, et al. Erdafitinib or chemotherapy in advanced or metastatic urothelial carcinoma. N Engl J Med. 2023. doi: 10.1056/NEJMoa2308849 3. Protocol for: Loriot Y, Matsubara N, Park SH, et al. Erdafitinib or chemotherapy in advanced or metastatic urothelial carcinoma. N Engl J Med. 2023. doi: 10.1056/NEJMoa2308849